Water models
Overview
Teaching: 15 min
Exercises: 0 minQuestions
Why do we want to use water in out simulations?
What water models are commonly used?
How to chose a model of water for a simulation?
Objectives
Learn how water molecules are modeled in simulations
Understand how choice of water model can affect a simulation
Introduction
The goal of bio-molecular simulations is the accurate and predictive computer simulation of the physical properties of biological molecules in their aqueous environments. There are two approaches to solvation: a suitable amount of explicit water molecules can be added to prepare a fully solvated simulation system. Alternatively, water can be treated as a continuous medium instead of individual molecules.
Realistic water environment is essential for accurate simulation of biological molecules.
Two approaches to solvation:
- Add explicit water molecules.
- Treat water as a continuous medium instead of individual molecules.
Continuum models
As continuum models do not add any non-bonded interactions to a simulation system, they are significantly faster than explicit solvation. Implicit water models, however, have limitations. They cannot reproduce the microscopic details of the protein–water interface. The conformational ensembles produced by continuum water models differ significantly from those produced by explicit solvent and cannot identify the native state of the protein [Ref]. In particular, salt bridges were over-stabilized, and a higher-than-native alpha helix population was observed. These models are still useful for example for calculation of binding free energies or for flexible protein-protein docking.
- Significantly faster than explicit solvation.
- Cannot reproduce the microscopic details of the protein–water interface.
- Do not produce same conformational ensembles as explicit water (salt bridges are over-stabilized, a higher-than-native alpha helix population).
- Useful for calculation of binding free energies or for flexible protein-protein docking.
Most molecular dynamics simulations are carried out with explicit water molecules.
Explicit models
Most molecular dynamics simulations are carried out with the solute surrounded by a droplet or periodic box of explicit water molecules. In a typical case, water molecules will account for over 80% of the particles in the simulation. Water–water interactions dominate the computational cost of such simulations, so the model used to describe the water needs to be fast as well as accurate.
- Typically water molecules account for over 80% of the particles in the simulation.
- Water–water interactions dominate the computational cost of such simulations.
- The water model needs to be fast and accurate.
Explicit water models are empirical models focused on reproducing a number of bulk properties in a particular phase. For example, some models reproduce well protein hydration energies, while others predict excellent water structure but not so good for hydration free energy. Most importantly, none of water models accurately reproduce all of the key properties of bulk water simultaneously. Simulators can be adversely affected by inaccurate water models in unpredictable ways.
- Explicit water models are empirical models derived to reproduce some bulk properties in a particular phase.
- None of water models accurately reproduce all of the key properties of bulk water.
- Which water model is optimal for the simulation depends on the desired properties of interest.
Therefore, the desired properties of interest must be considered when choosing a water model for a molecular simulation, since this will determine which model is most appropriate.
An early water model.

Water molecules have OH distance of 0.9572 \(\unicode{x212B}\) and HOH angle of 104.52°. Early water models used a rigid geometry closely matching that of actual water molecules. To achieve this O-H and H-H distances are constrained with harmonic potential.
Point charges in classical water models replace electron density distribution. They are meant to reproduce the electrostatic potential of a molecule. Therefore, they are usually derived by fitting to the electrostatic potential around a water molecule. There are some downsides for this approach, which we will discuss later.
- Rigid geometry closely matching actual water molecule.
- O-H and H-H distances are constrained with harmonic potential.
- Point charges replace electron density distribution.
How are water models derived?
A good water model must faithfully reproduce six bulk properties of water:
- Static dielectric constant, \(\epsilon_{0}\)
- Self diffusion coefficient, \(\vec{D}\)
- Heat of vaporization, \(\Delta{H}_{vap}\)
- Isobaric heat capacity, \(\vec{C}_{p}\)
- Thermal expansion coefficient, \(\alpha_{p}\)
- Isothermal compressibility, \(\kappa_{T}\)
Many water models with different level of complexity (the number of interaction points) have been developed. We will discuss only the models most widely used in simulations, and refer you to the excellent article in Wikipedia for an overview of all water models.
3-charge 3-point models.
These models have three interaction points corresponding to the atoms of the water molecule. Only oxygen atom has the Lennard-Jones parameters.
- Three interaction points corresponding to the atoms of the water molecule.
- Only oxygen atom has Lennard-Jones parameters.
- 3-site models are commonly used because computationally they are highly efficient.
| TIP3P (transferable intermolecular potential) $\circ$ Rigid geometry closely matching actual water molecule. |
 |
| SPC/E (simple point charge) $\circ$ More obtuse tetrahedral angle of 109.47°. $\circ$ Adds an average polarization correction to the potential energy function. $\circ$ Reproduces density and diffusion constant better than the original SPC model. |
 |
Models with three sites are commonly used because they are computationally efficient. The interactions between two molecules can be computed using only nine distances.
3-charge 4-point models.
In these models the negative charge is not centered on the oxygen atom, but shifted towards hydrogen atoms. This position is represented with the fourth dummy atom (EP) located near the oxygen along the bisector of the HOH angle. The idea is to improve representation of electrostatic potential without changing its geometric properties. The idea is to improve electrostatic potential representation without changing the geometric properties of the molecule.
- The negative charge is not centered on the oxygen atom, but shifted towards hydrogen atoms
- The position of charge is represented with the fourth dummy atom (EP)
- EP is located near the oxygen along the bisector of the HOH angle.
| TIP4P-Ew $\circ$ Improves association/dissociation balance compared to 3-point models. |
 |
Water models have their limitations.
It is important to understand the limitations of water models. Models are unable to reproduce quantitatively all characteristics of real water. When chosen appropriately for the problem, however, they can provide useful insight into water’s behavior.
- Early water models were developed with cut-off of electrostatic interactions. Using these models with full electrostatic method results in stronger electrostatic interactions and consequently higher density.
- Most of the more complex new water models attempt to reproduce specific properties of a specific phase, but this comes at the expense of other properties.
- TIP3P model predicts hydration free energies of small neutral molecules more accurately than the TIP4PEw model.
- 4-charge 5-point model TIP5P predicts excellent water structure, but poor hydration energies.
Challenges in developing water models.
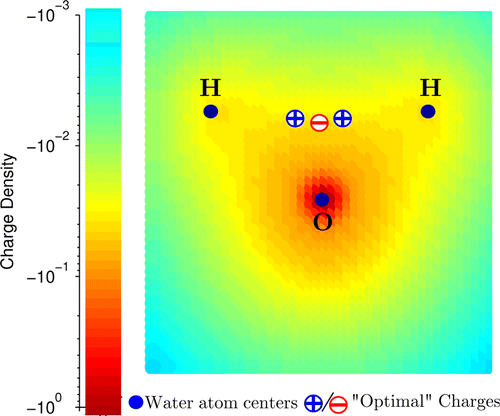
A key challenge in developing water models is to find an accurate yet simplified description of the charge distribution of the water molecule that can adequately account for the hydrogen bonding in the liquid phase.
Traditional approach is to place point charges on or near the nuclei. Afterwards, however, it was discovered that 3 point charges reproduce the electrostatic potential of water molecules significantly more accurately when they form tight clusters.
- Finding an accurate yet simplified description of the charge distribution that can adequately account for the hydrogen bonding in the liquid phase.
- Traditional approach is to place point charges on or near the nuclei.
- Electrostatic potential of water molecule is reproduced considerably more accurately with 3 point charges when they form tight cluster.
Optimal Point Charges (OPC and OPC3)

The last and the latest water models we will look at is the OPC (Optimal Point Charges). OPC [2] belongs to the family of 3 charge, 4 point models while OPC3 is 3 charge, 3 point version [4]. The key difference from the previous models is that it was designed without geometrical restraints. This design approach is based on the observation that QM electrostatic potential of water molecule is reproduced considerably more accurately with 3 point charges when they form tight cluster of the point charges away from the nuclei than the more traditional distribution with point charges placed on or near the nuclei.
- Removing point charge positioning restrictions allowed for considerably better reproduction of the six bulk properties of water.
- Designed without geometrical restraints.
- Considerably better reproduces the six bulk properties of water.
Polarizable water model OPC3-pol
Rigid models with fixed charges are far from perfect since they do not account for many of the physical effects of real water. Among these physical effects are many subtle quantum effects, water flexibility, and, most importantly, electronic polarizability. In the absence of polarizability, a model cannot respond to changes in polarity in its micro-environment, which is very relevant in many types of simulations. As an example, the dipole moment of a real water molecule changes by almost 40% when it crosses the water-vapor interface, whereas with a fixed charge model, the dipole moment does not change.
Polarization is usually achieved by adding model oscillators, referred to as Drude particles. The Drude particle is a massless virtual site with a partial electric charge attached to an atom by means of a harmonic spring. Simulations of classical Drude oscillator involve repositioning the Drude particle via a computationally expensive self-consistent procedure.
This cost can be reduced by assigning a small mass to each Drude particle, and evolving the simulation using the extended Lagrangian dynamics. Simulations using polarizable water models such as (iAMOEBA and SWM4-NDP) still take at least four times as long as those using rigid non-polarizable water. Additionally, Drude water models have the drawback of requiring matching, polarizable force fields for biomolecules.
OPC3-pol performs almost as well as non-polarizable water models and requires no specialized force fields for proteins or nucleic acids. For high computational efficiency, the OPC3-pol model treats the Drude particle as an “ordinary” atom in the molecular dynamics system: the water oxygen mass is split equally between the oxygen atom and the Drude particle.
The critical benefit of the approach is that OPC3-pol water model can run as fast as classical non-polarizable water models with the existing biomolecular force fields.
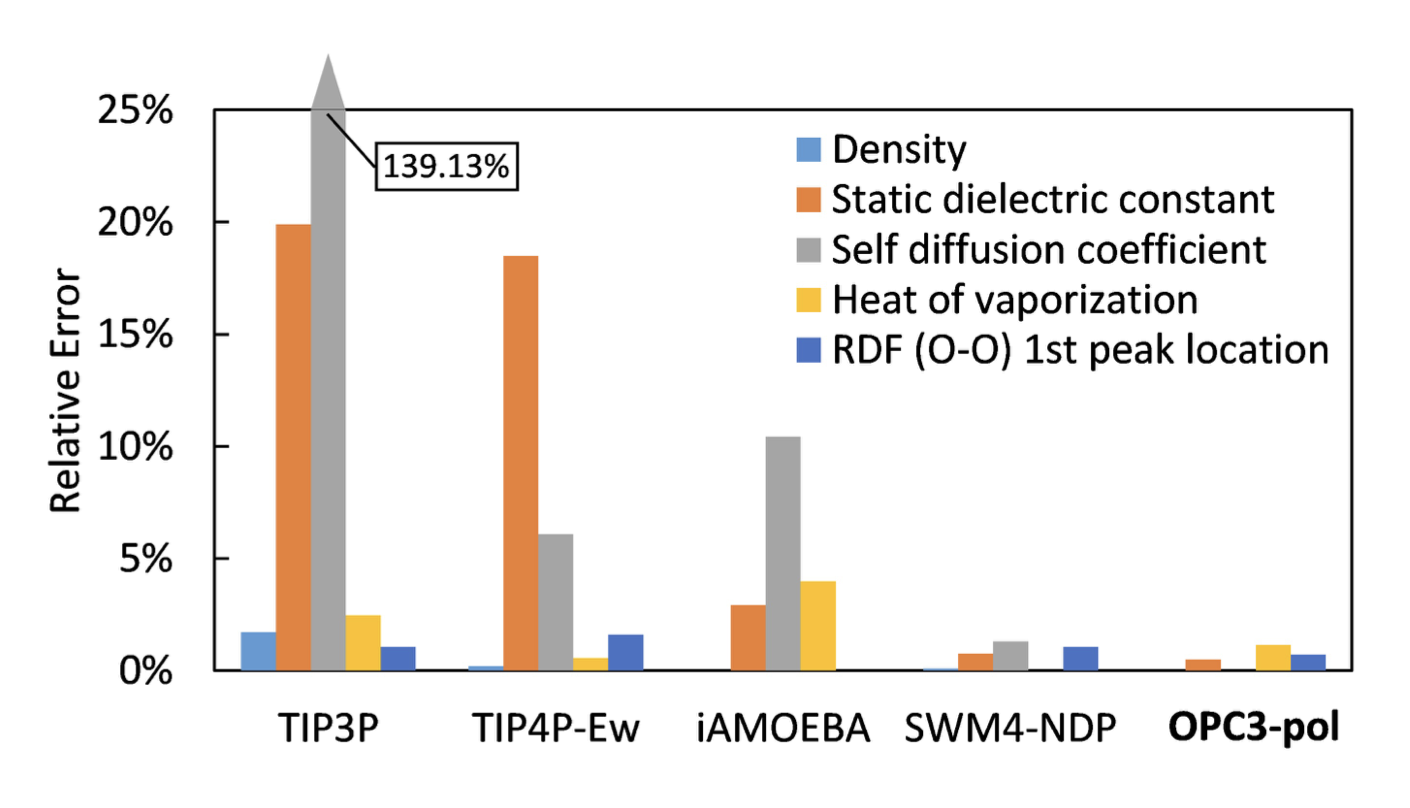
Quality of different water models
Let’s have a look at the quality scores of different water models summarized in the figure below. The figure shows how quality score depends on the dipole and quadrupole moments. Interestingly the test models in which the moments were close to the QM values had low quality. And the models that scored better had moments very different from the QM values.
- The test models in which the moments were close to the QM values had low quality.
- The models that scored better had moments very different from the QM moments.
This indicates that three point charges, even if placed optimally, are not enough to represent the complex charge distribution of real water molecule to the needed degree of accuracy.
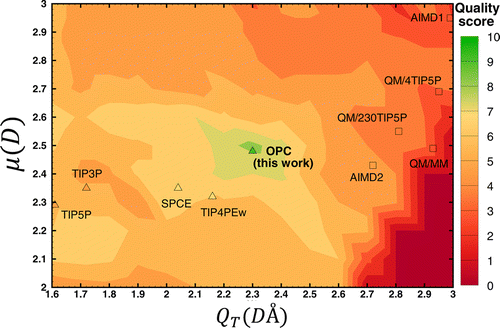
The distribution of quality scores for different water models in the space of dipole (μ) and quadruple (QT) moments. Figures from [2].
Performance Considerations
The time to compute interactions between a pair of water molecules is approximately proportional to the number of distances between each pair of interaction points. For the 3-point model, 9 distances are required for each pair of water molecules. For the 4-site model, 10 distances are required (every charged site with every charged site, plus the VDW O–O interaction).
- Computation cost is proportional to the number of pairwise distances.
- 3-charge 3-point model: 9 distances
- 3-charge 4-site model: 10 distances (3x3 Coulomb interactions plus one VDW O–O interaction).
Other things to consider
Water models in common use in bio-molecular simulation have traditionally only been parameterized for a single temperature of 298K (SPC/E, TIP3P, etc.)
Force Field Parameters of the common Water Models
| TIP3P | SPC/E | TIP4P-Ew | OPC | OPC3 | |
|---|---|---|---|---|---|
| OH | 0.9572 | 1.0 | 0.9572 | 0.8724 | 0.9789 |
| HH | 1.5136 | 1.63 | 1.5136 | 1.3712 | 1.5985 |
| HOH | 104.52 | 109.47 | 104.52 | 103.6 | 109.47 |
| OM | - | - | 0.125 | 0.1594 | - |
| A(12) | 582.0 | 629.4 | 656.1 | 865.1 | 667.9 |
| B(6) | 595.0 | 625.5 | 653.5 | 858.1 | |
| qO | −0.834 | −0.8476 | −1.04844 | −1.3582 | -0.8952 |
| qH | +0.417 | +0.4238 | +0.52422 | +0.6791 | +0.4476 |
Nonbonded OPC3: sigma=1.7814990, epsilon=0.163406 pending convert to A, B
- Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K
- Building Water Models: A Different Approach
- Effect of the Water Model in Simulations of Protein–Protein Recognition and Association
- Accuracy limit of rigid 3-point water models
- A fast polarizable water model for atomistic simulations
Key Points
Continuum models cannot reproduce the microscopic details of the protein–water interface
Water–water interactions dominate the computational cost of simulations
Good water model needs to be fast and accurate in reproduction of the bulk properties of water